Hemoglobin S Hastalığı
Belli ırksal kökeni olan bir çocukta orak hücreli anemiyi tanımak genellikle zor değildir. Hemoglobin S heterozigotlarını ve bileşik hemoglobinopatileri tetkik etmek daha zordur.
A.Orak Hücreli Anemi Taşıyıcılığı
Hemoglobin S'in heterozigot şekli (PPS) olan bu durum anemi ya da herhangi bir klinik hastalığa yol açmaz. Çok sık izlenen bir genetik anomali olup Amerika' da yaşayan siyah ırk mensuplarının %7-8 kadarında taşıyıcılık mevcuttur. Bazı Afrika ülkelerinde taşıyıcılık ihtimali daha da yüksek olup, talasemi ile bileşik heterozigotluk ortaya çıkma riski bariz bir şekilde artmıştır. Bu yüksek prevelansın orak hemoglobinin sıtmanın endemik olduğubölgelerde sağkalım avantajı yaratmasından kaynaklandığı düşünülmektedir. Bu durum orak hemoglobinin Afrika'nın 4 değişik bölgesinden köken aldığının gözlenmesi ile doğrulanmıştır.
Tek orak geni bireyin genel sağlık durumunu etkilemez .Yaşam süresi normaldir. Bu hastalar ağrılı kriz ya da organ hasarı geliştirmezler. Orak hücreli anemi taşıyıcılarında izlenebilen tek anormallik %3 kadar vakada gelişebilen rekürren, ağrısız hematüridir. Ayrıca, birkaç vaka takdimi şeklinde orak hücreli anemi krizi ve ağır egzersiz, dehidratasyon ya da çok düşük oksijen basıncı ortamlarında ani ölüm bildirilmiştir. Rutin eBe normaldir. Periferik kan yaymasında orak hücreler izlenmezler. Eritrosit ömründe belirgin bir azalma yoktur. Dolayısıyla retikülosit artışı, LDH yüksekliği, indirekt hiperbilirubinemi dahil olmak üzere hemolitik aneminin bulguları yoktur. Hemoglobin elektroforezi diagnostiktir. Hemoglobin S % 44-45 civarındadır.
B.Orak Hücreli Anemi
Hemoglobin S hastalığının homozigot (PSPS) şekli olan orak hücre li aneminin tanısı kolaydır. Amerika' da siyah ırka mahsus bir hastalıktır ve bireyin ailesinde ebeveynlerin ikisi de heterozigottur. Klinik hastalık hemen her zaman çocuk 6 aya yaklaştığı zaman ortaya çıkar. Hayatın ilk 6-12 haftasında yüksek hemogloin F düzeyi ağır intravasküler oraklaşmayı engeller.
1. Klasik semptomlar ve bulgular: Orak hücreaneminin klasik semptom ve bulguları intravasküler oraklaşma. küçük damar tıkanması ve doku infarktlarına bağlı rekürren, ağrılı krizlerdir. İnfarktüs için en yüksek risk altındaki organlar kemik iliği, böbrekler, dalak, akciğer ve beyindir. Küçük çocuklar el ve ayaktaki küçük kemiklerin ilik nekrozlarına bağlı akut daktilit tablosunda gelebilirler. Daha büyük çocuklar mükerrer kemik ve eklem ağrılarından ve dalak infarktüsüne bağlı karın ağrısından yakınabilirler. Dalak işlevlerinin bozulmasına bağlı olarak gelişen pnömokokal sepsis orak hücreli anemili çocuklarda önde gelen ölüm nedenidir. Çocuk büyüdükçe akciğer, kalp, beyin gibi hayati organ hasarları artar. Tedavi edilmeyen hastalarda sabit bir vasküler oklüzyon riski vardır ve sıklıkla bir inmeye ya da majör kardiyopulmoner hasara bağlı olarak dördüncü dekattan önce ölürler. Tedavi edilmeyen çocuklarda yıllık inme riski % 1O civarındadır. Bu risk kronik transfüzyon ile< %1 düzeyine indirilebilir. Bu gözlem sebebiyle 12-6 yaş arasındaki homozigot hemoglobin S hastalarının orta serebral ve internal karotid arterlerde artmış kan akım hızı yönünden 6 ayda bir transkraniyal doppler ile taranması gerektiği önerisini gündeme getirmiştir. Erişkin orak hücreli anemi hastalarının % 5- 30 kadarı tekrarlayan vasküler oklüzyonlara (akut göğüs sendromu) ve pulmoner fibrozise bağlı olarak pulmoner hipertansiyon geliştirirler. Bu hastaların ortalama sağkalım süreleri 2 yıl kadardır.
2. Hastalık şiddeti: Orak hücreli aneminin şiddeti hemoglobin S konsantrasyonu, hemoglobinin deoksijenasyon derecesi, intrasellüler hemoglobin konsantrasyonu ve hemoglobin F düzeyi gibi bir çok parametreye bağlıdır. Homozigot 13 s 13 s hücreleri hücre dışına doğru artmış potasyum-klor kotransportu ve eritrosit membranı büküldükten sonra kalsiyuma bağımlı (Gardos) potasyum ihraç kanalı aktivasyonuna bağlı olarak dehidrate olma eğilimindedir. Bu durum hemoglobin polimerizasyonunu hızlandırır. Ayrıca, hemoglobin SC ve CC hastalarında target hücreleri oluşma mekanizmasında da rol oynar.
Tam kan viskozitesi ve doku mlkrodolaşı-mırıın debisi deoksijenasyon düzeyi ve geri dönü-şümsüz orak hücre oluşumu ile korelasyon gösterir.Oksijenlerımiş orak hücreler normalin 1. 5 katı viskoziteye sahiptir. Deoksijenasyon ile birlikte viskozite normalin 1O katına yükselir. Viskozite artışı kısmen hastanın anemisi ile telafi edilir. Ancak oraklaşma ile birlikte kan akımı özellikle küçük arterioller düzeyinde belirgin olarak azalır. Belirgin dehidratasyonda olduğu gibi hastanın hematokritindeki herhangi bir artış viskoziteyi daha da artırarak hastalığın şiddetine katkıda bulunur.
Vazookluzif ataklarm sıklığı ve şiddeti ayrıca orak hücrelerin endotele yapışma eğilimini deyansıtır. Orak hücreler CD36, u4Pı integrin, sülfatlanmış glikolipid, fosfotidilserin maruziyeti ve Luteran kan grubu antijeni gibi birçok adezyon reseptörü eksprese ederler. Sitokin tarafından indüklenenendotelyal vasküler adezyon molekülü-1 (VCAM-1)ile von Willebrand faktörü ve trombospondin bağlayan uvP3 integrin orak hücre adezyonuna yardımcıolurlar. Trombosit adezyonu ve beyaz küre adezyon! aktivasyonu ile birlikte eritrositlerin endotel hücrelerine bağlanmaları okluzif atağın şiddetine katkıda bulunur. Hastalığın ilerlemesi ile birlikte doku hasarı kan akımının bozulmasına ve dolayısıyla hemoglobin S polimerizasyonunun ve orak hücre edezyonunun daha da artmasına yol açar. Son olarak intrasellüler hemoglobin F konsantrasyonu polimer oluşumunu fiziksel olarak engellemek suretiyle hastalığın şiddetini belirlemede majör bir rol oynar.
Orak hücreli anemi hastaları ayrıca ağır hemolitik anemi belirtileri de gösterirler. Laktat dehidrogenaz ve indirekt bilirubin düzeyleri artmıştır. Klinik sarılık da ortaya çıkabilir. Ani ve tekrarlayan apıastik kriz atakları riski vardır. Burada geçici bir kök hücre yetmezliği nedeniyle aneminin bir süreliğine ağırlaş-ması söz konusudur. Parvovirüs infeksiyonu apıastik krizin iyi bilinen nedenlerinden birisidir. Böbrekte majör hasar oluşmasına bağlı olarak eritropoietin üretiminin bozulması da bir neden olabilir. Eritroid serideki genişlemenin kemik yapısına etkisi çok çarpıcı olabilir. Distal kemiklerde aktif kemik yapımı gözlenebilir. Bazen buna kemik korteksinin incelmesi ve özellikle kafatasında olmak üzere kemikte yeniden yapılanma (= remodeling) eşlik edebilir. Frontal belirginleşme ağır hastalığın bir işaretidir.
3. Komplikasyonlar: Hafif-orta şiddette büyüme geriliği ve pubertede gecikme gözlenebilir. Bu durum çinko eksikliği ile ilgili olabilir. Ancak, hastalar 18- 21 yaş gibi normal fiziksel görünüm e ve cinsel olgunluğa ulaşırlar. Gebe kalma konusunda genellikle sorun yaşanmaz. Ancak, gebelik sırasında atakların sayısı ve şiddeti artabilir. Ergenlik ile birlikte genç erkeklerde tekrarlayan priapism atakları sıklıkla gelişebilir.
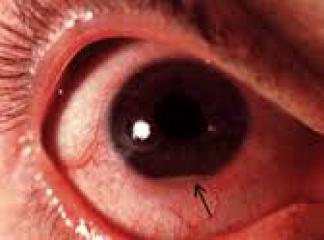
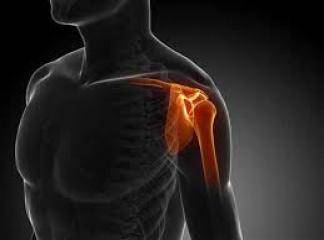
Orak hücreli anemının diğer komplikasyonları arasında proliferatif retinopati, safra kesesi taşları, femur ve humerus başlarının aseptik nekrozları, bacak ülserleri, böbrek yetmezliği ve akut göğüs sendromu vardır. Proliferatif retinopati ileri yaşlarda ortaya çıkar. Ancak, safra kesesi bilirubin pigment taşları genellikle 2. ya da 3. dekatta izlenirler. Malleol üstündeki cilt ve cilt altı yağ dokusunun infarktına bağlı zor iyileşen bacak ü1serleri genellikle 2. Ya da 3. dekatta ortaya çıkarlar. Femur ve humerus başlarnun aseptik nekrozları prostetik cerrahi gerektirebilen sakatlayıcı bir artrite yol açabilirler. Ayrıca aseptik nekrozlar pulmoner yağ embolisine yol açabilirler. Salmonella ve staphylococcus aureus osteomiyelitleri için risk faktörüdürler. Hayatın ilerleyen yıllarında böbrek yetmezliğine yol açabilen renal bozukluklar izlenebilir. Akut göğüs sendromu erişkin orak hücreli anemi hastalarında hayatı tehdit eden bir komplikasyondur. Hastalar ateş, öksürük, hipoksemi ve göğüs ağrısı ile gelirler. Genellikle viral ya da bakteriyel bir infeksiyon kanıtı yoktur. Göğüs filminde multilob infiltrat izlenir ki otopside bu görünümün karşılığı yaygın tromboz/yağ embolisidir. Akut göğüs sendromu belirgin hipoksi, solunum yetmezliği ve ölüm ile sonuçlanabilir. Sıklıkla hematokrit ve tam kan viskozitesinin ani artışı ile ilişkili olan multi organ yetmezliği ve inmeler de hayatı tehdir eden komplikasyonlardır.
4. Hastahğın seyri: Hastalığın seyrinde belirleyici faktörler vazookluzif atakların sıklığı, ane mini n şiddeti ve başka anormal hemoglobinlerin olup olmamasıdır. Hemoglobin C ve D hastalığın şiddetini arttırabilirler. Buna karşılık yüksek hücre içi hemoglobin F düzeyi daha hafif bir hastalık fenotipine yol açar. Herediter hemoglobin F perzistansı olan Ortadoğu ve Batı Afrika'lı hastalarda çok hafif bir hastalık şekli vardır. Orak hücreli anemi hastasında eşlik eden demir eksikliği ya da talasemi nedeniyle hücre içinde hemoglobin konsantrasyonunun azalması da oraklaşma eğilimini ve vazookluzif atak sıklığını azaltabilir.
C.Hemoglobin SC Hastalığı
Hemoglobin SC hastalığı psps hastalığından daha seyrektir. Klinikte orak hücreli anemi ile aynı belirtilerle seyredebilir. Ancak, vazookluzif sorunların sıklığı ve şiddeti daha azdır. Orak hücreli aneminin tersine dalak infarktı sık değildir. çoğu erişkin hemoglobin SC hastasının büyümüş ve kolayca palpe edilebilen dalakları vardır. Diğer organ hasarları da (renal medüller infarkt, santral sinir sistemi trombozu ve kardiyopulmoner tromboz) daha seyrektir. Hemoglobin SC hastalarında aşikar hematüri daha sıktır. Bayanlarda spontan abortus hızı çok yüksektir
Hastalığın laboratuar tanısı zor değildir. Hafiforta şiddette hemolitik anemi vardır. Eritrositler arasında hedef hücreler oranı oldukça artmıştır (Renkli resimler 14), Eritrositler içinde kristaloluşması, sferosit oluşması ve dolayısıyla hafif ortalama eritrosit hemoglobin konsantrasyonu (MCHC) artışı ve gerçek oraklaşma olmaması söz konusudur. HPLC her iki hemoglobini de hızlıca gösterebilir. Hemoglobin elektroforezi bulguları tanısaldır.
D.Hemoglobin S/ß-Talasemi
Hemoglobin S ve ß-talaseminin birlikte kalıtılması siyah ırkta ve Akdeniz toplumlarında sık izlenen diğer bir hastalıktır. Hemoglobin S Alrika-talasemi ile kombine olduğunda (hemoglobin S/ß+-talasemi) hastalık tablosu hafif olup orak hücreli anemi taşıyıcılığına benzer. Hafif-orta şiddette hemolitik anemi ve splenomegali gözlenebilir. Hemoglobin elektroforezinde hemoglobin A üretimi azalmıştır. Hemoglobin S/A dağılımı %60/40 şeklinde olup hemoglobin F ve Aı üretimine bağlı olarak daha da yüksek olabilir.Hemoglobin S'in daha hakim olması S/ß+ -talasemi hastalarını heterozigot S (orak hücreli anemi taşıyıcıları) hastalarından kolaylıkla ayırt ettirir.
Akdeniz toplumlarında hemoglobin S'in ßO-talasemi ile kombine olması (hemoglobin S/ßO-talasemi) daha ağır bir hastalık tablosuna yol açar. Hastalar dalak infarktı dışında orak hücreli anemi semptomlarının çoğunu ve hatta hepsini gösterirler. Bu hastalarda vazookluzif semptomların ağırlığı eritrositler içindeki hemoglobin F düzeyine bağlıdır. Hemoglobin F düzeyi %10-lS'in üstünde ve bütün eritrositiere eşit dağılmış olduğunda hastanın kliniği çok daha hafiftir. Hemoglobin F düşük olduğunda klinik tablo, yayma bulguları ve hemoglobin elektroforezi SS hastalığına benzer.
E.Diğer Bileşik Heterozigotlar
Hemoglobin D, O-Arab ve Lepore hemoglobin S ile birlikte sıklıkla kalıtılan diğer ß-globin zincir defektleridir. Hemoglobin SD ve S/O-Arab hastalıkları orak hücreli anemiye benzer ancak daha hafif klinik bozukluklara yol açarlar. Hemoglobin S/Lepore de daha hafif bir hastalıkla sonuçlanır.
İLGİNİZİ ÇEKEBİLİR