Huntington Hastalığı
Huntington hastalığı (HD) klinik olarak ilerleyen hareket bozuklukları ve demansla nitelenen otozomal baskın bir kalıtsal hastalıktır, striatum (kaudate ve putamen) dejenerasyonu olur. Hareket bozukluğu tüm beden bölgelerini etkileyen kesik, hiperkinetik, bazen diskinetik hareketler (korea) görülür; hastalarda parkinsonizmle birlikte bradikinezi ve katılık (rijidite) gelişebilir. Hastalık kesintisiz şekilde ilerler, ortalama seyir olan yaklaşık 15 yıl sonra ölümle sonuçlanır. HD olan bütün hastalarda bir tip mutasyon vardır bir trinükleotid tekrarlama ekspansiyonudur, 4p16.3'te yerleşik ve büyük bir proteini (huntingtin) kodlayan gendir. Gendeki polimorfik CAG trinükleotid tekran proteindeki bir poliglutamin yolağını kodlamaktadır.
Normal alelerde 11 ile 34 kopya tekran olur; hastalık yapan alellerde tekrar sayısı artmıştır, bazen yüzlere kadar çıkar. Tekrar sayısı ne kadar fazlaysa hastalığın o kadar erken dönemde ortaya çıkacağına yönelik güçlü bir genotipfenotip korelasyonu bulunmaktadır. Bununla birlikte, belirtiler başladıktan sonra hastalığın seyri tekrar uzunluğuna anlamlı şekilde bağımlı değildir. Spermatogenez sırasında tekrar ekspansiyonlar olur ve paternal geçişe bir sonraki kuşakta hastalığın erken dönemde başlaması eşlik eder. Yeni gelişen mutasyonlar nadirdir ve en belirgin olarak "sporadik" olgular paternal tanımlamalardaki hatalarla ya da hastalığın ortaya çıkması öncesinde ebeveynin ölmesiyle ilişkili olabilir.
Hastalığı olmayan bazı babalardaki tekrar ekspansiyonlar çocuklarına aktarım sırasında daha da ekspansiyona uğrar. Hastalıklarının belirti verme aşamasının öncesinde olan bireylerin tanımlanması elbette ki yoğun bir etik yük taşır ve uygun danışmanlık hizmetinin yokluğunda yapılmamalıdır. Proglutamin yolağı ekspansiyonu olan huntingtin proteininde hastalığın nasıl geliştiği henüz net olarak bilinmemektedir. Bir hipotez proglutamin ekspansiyonu olan mutant huntingtin'ın uzantılarıyla çeşitli transkripsiyon faktörlerine bağlanıp bunları sekestre ederek kritik proteinlerin sentezini azalttığını savunmaktadır. Tamamen olmasa da alternatif bir görüş mutant huntingtin'ın mitokondrilerde nöro dejenerasyona yol açan işlevsel anormaliklere neden olduğu olasılığıdır. Bu işlevsel anormalliklerin bir kısmı aslında mitokondrideki elektron aktarımında rol oynayan proteinlerin ve antioksidan proteinlerin sentez ve transkripsiyonunda azalmadan kaynaklanabilir. Protein bedende yaygın şekilde ifade edildiğinden beyin bölgelerinde tutulumun neden bu kadar kısıtlı olduğu net olarak bilinmemektedir. Anormal huntingtin birikebilir ve protein birikimleri dokuda gözlenebilir. Anormal proteinin uygun şekilde katlanamaması ve bu hatalı katlanmış proteinin bazı nöronlarda apoptozu tetiklemesi olasıdır. Bununla birlikte, patogenezin bu mekanizması henüz kanıtlanmamıştır.
Morfoloji
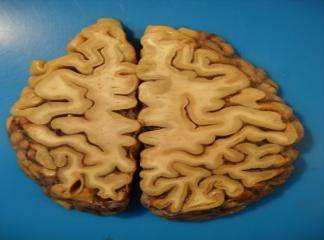
Makroskopik incelemede beyin küçüktür ve kaudate çekirdekte ve bazen daha az dramatik olarak putamende belirgin atrofi vardır. Patolojik değişiklikler hastalığın seyri sırasında kaudat‘ta medialdan laterale doğru, putamende ise dorsalden ventrale doğru ilerler. Globus pallidusta ikincil atrofi olabilir ve lateral ile üçüncü ventriküllerde genişleme vardır. Atrofi sıklıkla frontal lobca, daha az sıklıkta parietallobta ve nadiren korteksin tamamında da görülebilir. Mikroskopik incelemede bu striatum bölgelerindeki ağır nöron kaybı göze çarpar. Küçük nöronların kaybı sıklıkla büyüklerinin kaybından önce gelir. Enkefalin, dinorfin ve P maddesiyle birlikte nörotransmitter olarak yaminobutirik asit kullanan orta boyuttaki dikenli nöronlar özellikle etkilenir. Nöron kaybına karşı her zamanki tepkimede görülenden daha kapsamlı bir fibriler gliozis gözlenir. Striatumdaki dejenerasyonun derecesi ile motor belirtilerin şiddeti arasında doğrudan bir ilişki vardır; kortikal nöron kaybı ile demans arasında ilişki bulunmakla birlikte daha düşük düzeydedir. Geri kalan striatal nöronlarda ve kortekste ubikuitinlenmiş huntingtin proteininin birikimlerini içeren çekirdek içi inklüzyonlar vardır.
Klinik Özellikler
Hastalık ortaya çıktığında hasta sıklıkla 40'lı ve 50'li yaşlardır ve CAG tekrarının uzunluğuyla ilgilidir. Tekrar uzunluğu 70 kopyayı aştığında hastalık ergenlikte vefatta daha da erken dönemde ortaya çıkabilir, juvenil HD adı verilir. Motor belirtiler sıklıkla bilişsel bozulmadan önce ortaya çıkar. HD' de hareket bozukluğu kore biçimindedir, tüm beden bölgelerinde istemsiz kesintili hareketler vardır; ekstrem itelerdeki kıvrımlı hareketler tipiktir. Daha yüksek kortikal işlev bozukluğunun erken belirtileri arasında unutkanlık ile dir şünce ve affekt bozuklukları yer almaktadır ve ağır demansa ilerleme olabilir. HD hastalarında intihar riski yüksektir, en sık rastlanan doğal ölüm nedeni eklenen enfeksiyonlardır.
İLGİNİZİ ÇEKEBİLİR