Fibrozise Yol Açan Hastalıklar
İdiopatik Pulmoner Fibrozis
Kriptojenik fibrosing alveolit olarak da bilinen idiopatik pulmoner fibrozis (İPF), histolojik olarak diffüz interstisiyel fibrozis ile karakterize, nedeni bilinmeyen bir pulmoner hastalıktır. Bu hastalık ileri evrelerinde şiddetli hipoksi ve siyanıza neden olur. İPF' de tekrarlayan alveolitti başlatan ajan bilinmemektedir. Erkeklerde kadınlardan daha fazla görülür. Hastaların üçte ikisi kadarı hastalık başladığında 60 yaşın üzerindedir. İPF tanısı için Ussal interstisiyel pnömoni olarak adlandırılan histolojik fibrozis patterninin görülmesi gerekir. Ancak, benzer histolojik pattern asbestozis, kollajen vasküler hastalıklar gibi iyi tanımlanmış antiteler ve birçok diğer durumlarda da görülebilir. Bu nedenle "idiopatik" demeden önce bu patterne yol açabilecek bilinen tüm nedenlerin ekarte edilmesi gereklidir.
Morfoloji
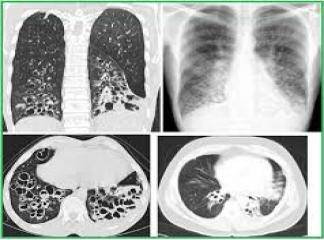
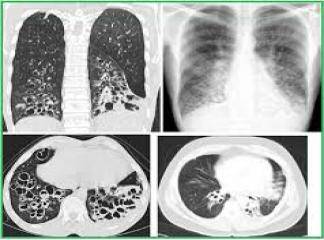
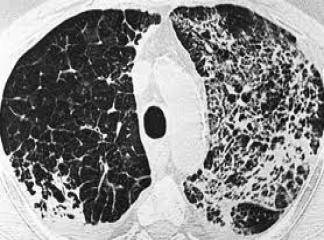
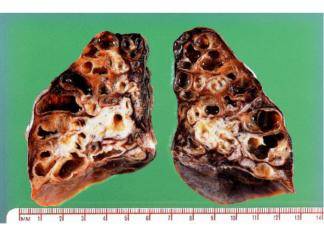
İnterlobüler saptalar boyunca karlı dokunun içe çekilmesi nedeni ile plevral yüzeylerde makroskopik olarak kaldırım taşı görünümü vardır. Kesit yüzünde belirgin şekilde alt loblarda ve subplevral bölgelerde ve interlobüler saptalar boyunca yerleşen lastiğimsi, sert, beyaz fibrotik alanlar görülür. İF‘de izlenen fibrozis patternine ussal interstisiyel pnömoni (İp) denilir. İp patterninde dağî-şen derecelerde ve zamanla da değişiklik gösteren odaksal interstisiyel fibrozis görülür. En erken lezyon fibroblastik fokum olarak görünen fibroblast proliferasyonunu içerir. Zamanla bu alanlar daha az hücresel eleman içerir ve daha fazla kollajeniz görünürler.
Oldukça tipik olarak erken ve geç evre lezyonların aynı anda birlikte görüldüğü heterojen bir görüntü (temposal heterojenice) bulunur. Yoğun fibrozis alveol duvarlarının kollarsına ve Hiperplastik tip i pnömositler ya da bronşiyol epiteli ile döşeli kıstık boşluklar oluşmasına neden olur (bal peteği fibrozis). İnterstisiyel inflamasyon genellikle odaksaldır ve alveol sepyalarını infiltre eden çoğunluğu lenfosit, arada plazmosit, mast hücresi ve eozinofillerden oluşur. Sekonder pulmoner hipertan sif değişiklikler (pulmoner arter duvarında intihal fibrozis ve medikal hipertrofi) sıklıkla izlenir.
Klinik Seyir
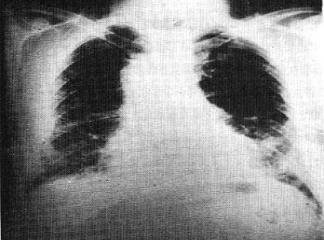
İPF genellikle sinsi başlar ve giderek artan balgam çıkarmayan öksürük ve ilerleyici dispne görülür. Fizik muayenede İF‘li hastaların çoğunda inspirasyonda karekteristik "kuru" ya da "Velcro benzeri" (yapışkan bir bandı açılırken çıkarttığı çıtırtı sesi, manşon rabli) çıtırtılar duyulur. Hastalığın daha ileri evrelerinde siyanız, kor pulmonale ve periferik ödem gelişebilir. İPF tanısını koymak ve pulmoner fibrozisin diğer nedenlerini ekarte etmek için cerrahi akciğer biyopsisi altın standarttır. Maalesef İPF tedaviye rağmen ilerler ve ortalama sağ kalım 3 yıl ya da daha azdır. Tek kesin tedavi şekli akciğer transplantasyonudur.
İLGİNİZİ ÇEKEBİLİR