Di George Anomalisi Nedir
Di George Anomalisi Nedir
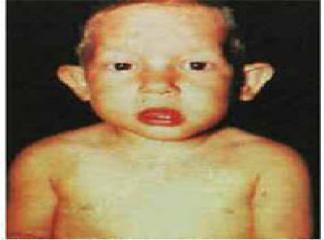
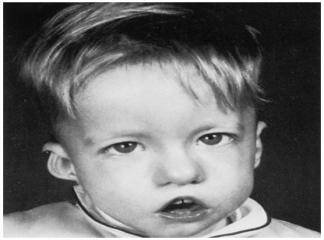
DiGeorge Sendromu (DGS), timus ve paratiroid bezinin konjenital yokluk sendromu olarak 1959‘da bildirilmifltir. A.M. DiGeorge (1) 1965 yılında immünitede timusun önemli bir rolü oldu¤unugöstererek, hipoparatiroidizm, timik hipoplazi ve tekrarlayan enfeksiyon geçiren olgularını bildirmifl, böylece timik aplazi ve konjenital hipoparatiroidizm birlikteli¤i ‘DiGeorge Sendromu‘ olarakadlandırılmıfltır. La Chapelle (2) 1981‘de bir ailenin dört etkilenen
bireyinde 22. kromozom translokasyonunu göstererek bu sendromun genetik orjini için ilk ipuçlarını sa¤lamıfltır.
Hastalýk çoðu olguda aðýr seyreder ve %75 olgu neonatal dönemde genellikle mevcut konjenital kalp hastalýðýnýn etiyolojik taramasý esnasýnda taný alýr. Ancak bazý geç bulgu veren ve kýsmen hafif seyreden klinik vakalarda hastalýðýn tanýsý okul çaðýna kadar gecikebilir. Bu ileri yaþta taný alan olgularda temel bulgularýn yaný sýra geliþme bozukluklarý, davranýþ bozukluklarý ve sýk tekrarlayan solunum yolu enfeksiyonlarýna baðlý komplikasyonlarda tabloya eþlik eder.
Hastalýðýn patogenezinde etken böigede (22q11.2) yer alan ve Di George sendromunda sýklýkla delesyona uðradýðý bilinen genlerden biri Di George Syndrome critical gene DGS geni yada bir baþka deyiþle Tbx1 genidir. Tbx1 gen ürünü, açýk yazýlýmýyla T-Box 1, bir transkripsiyon faktörü olarak aslýnda kontrolü altýnda tuttuðu birçok genin fonksiyonunu modifiye etmektedir. Ancak Tbx1 in bilhassa timus bezinin normal geliþimi için gerekli olduðu bilinmektedir. Di George sendromu hastalarýnda bu genin silinmesi sonucu timus yetmezliði geliþtiði ve bu sebeple T-hücrelerine baðlý immün cevabýn hastalarda hiçbir zaman tam oluþamamasýndan dolayý tedavi edilmesi zor aðýr enfeksiyonlar görüldüðü düþünülmektedir.
Sonunda Di George sendromu tanýsý alan bazý vakalarýn gebelik esnasýnda aþýrý alkol alýmýna atýf edildiði bilinmektedir. Ayrýca otozomal dominant geçiþ gösteren bir hastalýk olan (Damak-Kalp-Yüz) Velo-Cardio-Facial (VCF) sendromu, veya bir baþka adýyla Shprintzen sendromu, %70 olguda 22q11 bölgesinde bir delesyona baðlý geliþmekte ve Di George sendromu ile uyuþan bulgular verebilmektedir. Moleküler genetik mekanizma her iki sendromda da biribirine çok yakýn yerleþik ve bazen örtüþen genlere atýf edilmektedir. Ancak VCF sendromunu Di George sendromundan ayýran temel bulgular yarýk damak-dudak tutulumu, geliþme geriliði ve öðrenme zorluðu ile bereber seyretmesi ve kalýtsal geçiþ göstermesidir. Bu sebeple Di George sendromunun ayýrýcý tanýsýnda fetal alkol sendromu ve VCF sendromu unutulmamalýdýr.
Di George sendromunda ve VCF sendromu hastalarýnýn büyük çoðunluðunda moleküler genetik taný bir sitogenetik test yöntemi olan FISH (Fluorescent in-situ hybridization) tekniði ile 22. kromozomdaki delesyonun saptanmasý ile konur (22q11). Ancak interfaz (quick) FISH ile delesyonun saptanamasý halinde metafaz yaymada 22. kromozomu içeren bir translokasyonun taranmasý gerekebilir. Di George sendromu sýklýkla de-novo geliþen bir anomaliye baðlý olduðundan, bu sendromunda prrdiktif analiz veya taþýyýcýlýk testlernin yeri yoktur. Ancak hasta çocuk sahibi ebeveynlerin mükerrer gebeliklerinde FISH yöntemi ile kromozomal delesyon analizi prenatal taný amacýyla amniosentez yoluyla alýnan fetal hücreler üzerinde de uygulanabilir. VCF sendromunda ise tanýnýn atlanmasý ve taný konan ailelere danýþmanlýk hizmeti verilmesi koruyucu hekimlik açýsýndan zorunludur. Bu sebeple yarýk damak ve./veya konjenital kalp hastalýðý bulunan çocuklarda genetik etiyoloji taramasý yapýlmasý bir zorunluluktur.
İLGİNİZİ ÇEKEBİLİR