Di George Anomalisi
Di George Sendromu
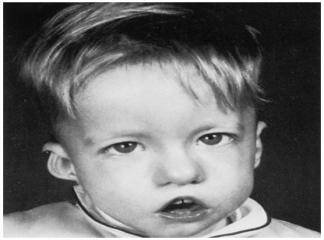
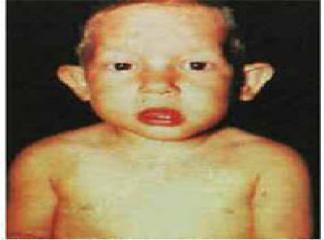
Di George sendromu yaklaşýk her 4,000 canlý doğumda bir (1:4000) olmak üzere oldukça nadir görülen bir konjenital hastalýktýr. Klinik olarak hastalýğýn semptomlarý hastadan hastaya bariz değişkenlikler ğöstermekle beraber bu hastalarda tipik bir yüz ifadesinin yaný sýra sýklýkla konjenital kalp defektleri, hipoparatiroidizim ve tekrarlayan ağýr enfeksiyonlar görülür.
Di George sendromu mayoz bölünme esnasýnda gamet hücrelerinde meydana gelen bir rekombinasyon hatasý sonucu 22. (yirim ikinci) kromozomdan geniş bir bölgenin silinmesi (delesyonu) yada translokasyonu (başka bir kromozoma taşýnmasý) sonucu de novo oluşan bir genetik anomalidir. Hastalýk genellikle kromozomdan geniş bir bölgenin kaybý sonucu oluştuğundan dolayý Di George sendromunda moleküler genetik patoloji tek bir gene indirgenemez. Dahasý bu hastalarda kromozomdan silinen bölgenin genişliği hastadan hastaya farklýlýk gösterdiğinden dolayý, vakalarda klinik tablonun delesyondan etkilenen genlerin sayýsý ile doğru orantýlý olarak ağýrlaştýğý bilinmektedir.
Di George sendronunda delesyona uğrayan bölgedeki genlerin embriyonun gelişimi esnasýnda 3. ve 4. faranjial keseciklerin normal gelişimi için gerekli olduklarý bilinmektedir. Dolayýsýyla bu embryonik yapýlardan türeyen organlar: timus bezi, paratiroid bezleri, aort yayý, dudak ve kulaklarýn alt kýsýmlarý Di George sendromunda tutulan primer anatomik yapýlardýr. Hastalarda timus tutulumu immün yetmezlik, paratiroid tutulumu kalsiyum metabolizma bozukluklarý, aort tutulumu konjenital kalp hastalýğý ve dudak-kulak tutulumu tipik yüz ifadesi olarak tabloya yansýr.
İLGİNİZİ ÇEKEBİLİR